Document de principes
L’évaluation de l’enfant ayant un retard global du développement ou un handicap intellectuel

Affichage : le 16 août 2018
Auteur(s) principal(aux)
Stacey A. Bélanger, Joannie Caron; Société canadienne de pédiatrie, Comité de la santé mentale et des troubles du développement
Paediatr Child Health 2018, 23(6):411–419. 
Résumé
Le retard global du développement (RGD) et le handicap intellectuel (HI) font partie des problèmes courants en milieu pédiatrique. Leur étiologie est très hétérogène. L’American Academy of Pediatrics, l’American Academy of Neurology et le protocole Treatable Intellectual Disability Endeavor (TIDE) de la Colombie-Britannique préconisent des explorations du RGD et du HI en plusieurs étapes, afin d’orienter les médecins vers une recherche étiologique qui optimise le rendement thérapeutique. Le présent document de principes propose un cadre pour l’exploration clinique du RGD et du HI chez les enfants, de même qu’une mise à jour du protocole d’exploration étiologique que peuvent suivre les médecins canadiens. Le protocole révisé repose sur les connaissances à jour et les lignes directrices en place. Les principaux éléments de l’exploration comprennent des tests de la vision et de l’ouïe en bonne et due forme, l’analyse chromosomique sur micropuce, le test d’ADN du gène de l’X fragile et les tests de niveau 1 de dépistage des erreurs innées du métabolisme traitables. En cas de manifestations neurologiques particulières, l’imagerie cérébrale est recommandée.
Mots-clés : Chromosome microarray; Global developmental delay; Intellectual disability; IEM
Le retard global du développement (RGD) et le handicap intellectuel (HI) touchent jusqu’à 3 % de la population pédia trique [1][2]. Le diagnostic de RGD est réservé aux enfants de moins de cinq ans, qui finissent souvent par répondre aux critères diagnostiques de HI et représentent probablement la même population (tableau 1). Puisque les diagnostics étiologiques de RGD et de HI se recoupent, il est naturel que les explorations en vue de poser un diagnostic définitif soient semblables. On ne soulignera jamais assez l’importance d’un dépistage précoce pour entreprendre les services de réadaptation et le traitement le plus rapidement possible. On parvient à établir l’étiologie du RGD et du HI dans de nombreux cas (de 40 % à 80 %) [3]. Il est donc essentiel que les pédiatres généraux du Canada, en collaboration avec des surspécialistes, adoptent une approche intégrative pour coordonner l’évaluation étiologique de cette population de patients.
| Tableau 1. Critères diagnostiques |
Retard global du développement
Diagnostic réservé aux enfants de moins de cinq ans Handicap intellectuel (trouble du développement intellectuel)*
|
|
Données tirées des références [1] et [2] * Les différents niveaux de sévérité sont définis sur la base du fonctionnement adaptatif et non plus sur la note au quotient intellectuel (QI) [1]. |
Le diagnostic est capital, car il permet d’agir à plusieurs égards [2] :
- L’adoption rapide d’un traitement causal ou de mesures de soutien
- La prévention des complications
- Un pronostic plus exact
- Des conseils génétiques précis sur le risque de récurrence et le diagnostic prénatal ou préimplantation, si la situation le justifie
- Un meilleur accès aux services communautaires
- La fin de la recherche d’un diagnostic ou (mieux encore) l’évitement de tests inappropriés, coûteux ou traumatisants
Le présent document de principes fournit un cadre pour l’exploration étiologique du RGD et du HI chez les enfants, afin d’aider les cliniciens à adopter des directives fondées sur des données probantes. Les auteurs proposent également une approche graduelle adaptée à la pratique clinique au Canada, au contexte clinique et aux ressources locales.
L’ÉTIOLOGIE DU RETARD DE DÉVELOPPEMENT GLOBAL ET DU HANDICAP INTELLECTUEL
La probabilité de parvenir à un diagnostic étiologique varie selon les études, le type d’exploration et la gravité du RGD ou du HI. Ainsi, la cause de HI grave (tel que ce terme est défini dans le DSM-5) était établie jusque dans 80 % des cas [4][5], mais celle des cas de HI léger n’atteignait qu’environ 24 % des cas [6]. Les catégories étiologiques et la proportion des diagnostics les plus courants sont présentées au tableau 2.
| Tableau 2. Causes de retard global du développement et de handicap intellectuel | ||
| Catégorie | Causes possibles | Proportion des diagnostics* |
| Facteurs prénatals intrinsèques | Problèmes génétiques Malformations du système nerveux central Problèmes métaboliques |
Jusqu’à 47 % Jusqu’à 28 % |
| Facteurs prénatals extrinsèques | Tératogènes et toxines (substances psychoactives, médicaments, etc.) Infections |
Jusqu’à 21 % |
| Facteurs périnatals | Asphyxie Prématurité Complications néonatales |
Jusqu’à 55 % |
| Facteurs postnatals | Négligence et environnement psychosocial Infections Traumatisme Toxines |
Jusqu’à 11 % |
|
Données traduites de la référence [3] * Pourcentage du total de cas de retard global du développement et de handicap intellectuel dont le diagnostic étiologique est établi et qui s’applique à ce groupe particulier. |
||
L’exploration étiologique
Les algorithmes recommandés par l’American Academy of Pediatrics (AAP) [2], l’American Academy of Neurology (AAN) [4] et le protocole du Treatable Intellectual Disability Endeavour (TIDE) [5] visent à simplifier l’exploration du RGD et du HI en limitant le nombre de tests chronophages ou sans utilité clinique et en favorisant l’utilisation efficace de ressources de santé restreintes.
Chaque algorithme est conçu pour dépister d’abord les étiologies les plus courantes ou traitables. En revanche, d’autres voies privilégient une approche qui repose sur des listes de vérification et des modèles de rapports de vraisemblance. Le clinicien met un terme aux explorations lors qu’il a l’impression qu’ils ne donneront rien de plus, même s’il n’a pas pu poser de diagnostic [3]. Une importante « perle clinique » consiste à rechercher les caractéristiques cliniques évocatrices d’une étiologie précise, puis à voir d’abord si des tests confirment le diagnostic. Lorsqu’il est impossible de dégager une cause apparente, il est recommandé d’opter pour une approche graduelle qui, de préférence, sera supervisée par un pédiatre en collaboration avec un généticien. Une telle approche de l’exploration est suggérée à la figure 1.
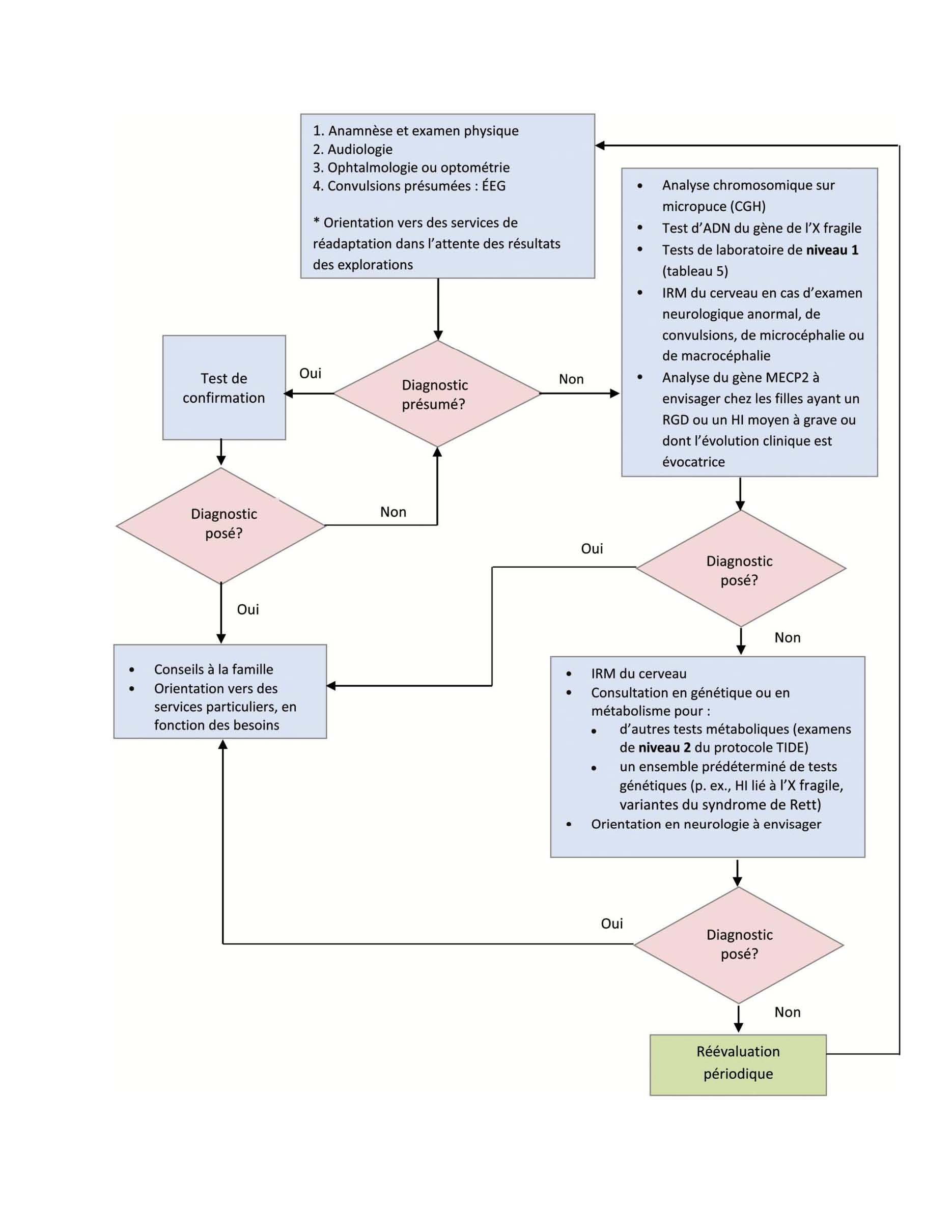
L’anamnèse et l’examen physique
Dans une récente analyse, les cliniciens ont posé un diagnostic étiologique fondé sur l’anamnèse et l’examen physique dans 12,5 % à 38,6 % des cas [3], ce qui confirme l’importance cruciale de ces éléments dans l’exploration [2][3][7][8]. Avec le plus de précision possible, les cliniciens doivent consigner l’histoire familiale sur trois générations, l’histoire psychosociale, l’histoire détaillée de la période prénatale et de l’accouchement et le moment des principales étapes du développement (tableau 3). Une évaluation neurodéveloppementale, qui inclut le niveau de développement et un examen physique systématique (tableau 3), peut faire ressortir un diagnostic précis ou orienter les tests de laboratoire. Lorsqu’on présume une étiologie spécifique ou qu’un trouble associé au RGD et au HI est bien établi dans l’histoire familiale, il faut d’abord demander des tests de dépistage visant le trouble en question (figure 1).
L’évaluation sensorielle
D’après l’AAN [4] et d’autres analyses [5][7][9], les enfants qui présentent un RGD et un HI doivent subir un examen de la vision (en optométrie ou en ophtalmologie) et de l’ouïe en bonne et due forme. Le dépistage d’un déficit visuel ou auditif peut modifier la prise en charge et susciter d’autres explorations.
Les tests génétiques
L’analyse chromosomique sur micropuce
L’AAP, l’AAN, l’International Standard Cytogenetic Array et l’American College of Medical Genetics (ACMG) cautionnent l’utilisation de l’analyse chromosomique sur micropuce (ACM, également appelée hybridation génomique comparative, ou CGH) en première ligne chez les enfants ayant un RGD ou un HI [2][4][9][10]. C’est le test qui, utilisé seul, donne les meilleurs résultats diagnostiques [7][8] (entre 8 % et 20 %). Seule l’éva luation clinique d’un clinicien chevronné spécialisé en RGD et en HI est plus efficace [2][4][11]. La variation du rendement observée dans les diverses études peut s’expliquer par l’absence de stratification pour établir la gravité du RGD et du HI et par la présence d’autres anomalies. Par conséquent, on ne sait pas si l’ACM est utile dans les cas de HI familiaux légers (selon le DSM-5). Ces patients ne représentent peut-être que les percentiles les plus bas de la courbe de quotient intellectuel de Gauss, et les étiologies sont souvent multifactorielles. En présence d’anomalies congénitales multiples, l’ACMG continue de recommander l’ACM en première ligne, à moins qu’un diagnostic précis soit envisagé [10].
Le caryotypage
Le caryotypage standard n’est pas recommandé en première ligne, car sa sensibilité correspond à moins de la moitié de celle de l’ACM chez les enfants ayant un diagnostic de RGD ou de HI. La résolution de l’analyse chromosomique traditionnelle est de 5 Mb à 10 Mb, alors que celle de l’ACM est de 0,05 Mb à 0,1 Mb. Cependant, on recommande le caryotypage plutôt que l’ACM en cas de présomption clinique d’aneuploïdie (p. ex., syndrome de Turner, trisomie 21) ou d’histoire familiale de réarrangements chromosomiques ou de multiples avortements spontanés [4][12]. Devant ce dernier scénario, il faut d’abord demander le caryotypage des parents.
Le test d’ADN du gène de l’X fragile
Le syndrome de l’X fragile est la principale cause génétique de HI chez l’enfant. Il touche de 2 % à 6 % des garçons et de 1 % à 4 % des filles ayant un HI. Puisque son phénotype clinique est souvent non spécifique chez les nourrissons et les jeunes enfants, l’AAP et l’AAN recommandent toutes deux d’envisager le test d’ADN du gène de l’X fragile (FMR1) en première ligne chez les garçons et les filles ayant un RGD ou un HI, tels que ces termes sont définis dans le DSM-5 [1][2][4][9][12][13]. Il est possible d’effectuer un ensemble prédéterminé de tests pour le HI lié à l’X fragile, mais on ne les envisage que dans les familles où au moins deux garçons sont atteints, sous la supervision d’un généticien [2].
Le test de dépistage du syndrome de Rett
Le syndrome de Rett est diagnostiqué chez 1,5 % des filles ayant un HI moyen à grave [2]. Selon l’AAP et l’AAN, le clinicien doit demander une analyse moléculaire du gène MECP2 lorsqu’il observe la symptomatologie caractéristique (développement normal au départ, suivi d’une perte de la parole et de l’utilisation volontaire des mains, stéréotypie des mains, anomalies de la démarche) ou que la fillette présente une atteinte intellectuelle moyenne à grave [2][4].
Le séquençage de l’exome ou du génome entier
Le séquençage de l’exome entier permet d’analyser les régions codantes de gènes connus et de déterminer les mutations causales chez jusqu’à 40 % des patients ayant un HI grave [14]. Dans certaines régions du Canada, cette technique relativement nouvelle devient accessible en clinique à faible coût. Des variations dont on ne connaît pas la signification demeurent problématiques et doivent être interprétées avec prudence. En raison de ces limites, le séquençage de l’exome ou du génome entier n’est pas recommandé en première ligne, mais pourrait l’être dans un avenir rapproché. Le Collège canadien des généticiens médicaux recommande à ses membres d’y recourir dans les cas de HI moyen à grave ou dans les cas syndromiques [15].
Le bilan métabolique
Les signaux évocateurs d’erreurs innées du métabolisme (EIM) sont énumérés au tableau 4. Ces observations justifient un bilan métabolique, mais certaines EIM sont beaucoup moins évidentes [5]. En 2011, l’AAN a recommandé d’effectuer des analyses métaboliques seulement en cas de forte présomption clinique, en l’absence de dépistage néonatal ou après l’exécution de tests génétiques et d’examens de neuro-imagerie qui n’ont pas permis de poser un diagnostic [4]. Au Canada, il n’y a pas d’ensemble prédéterminé et universel de tests de dépistage des troubles héréditaires chez les nouveau-nés; les programmes de dépistage néonatal varient selon les provinces et les territoires. Malgré un programme de dépistage efficace, il est tout de même facile de passer à côté de certaines EIM [2][5].
| Tableau 4. Signes évocateurs d’erreurs innées du métabolisme |
|
Il serait idéal de pouvoir rencontrer rapidement un généticien clinique ou un spécialiste du métabolisme afin d’obtenir une évaluation de l’EIM la plus probable, mais dans la majeure partie du Canada, ces services ne sont pas accessibles. Il est également frappant de constater que jusqu’au deux tiers des enfants ayant un RGD ou un HI ne présentent pas de constellations de symptômes caractéristiques qui orienteraient le diagnostic. En raison de la non-spécificité des symptômes, les cliniciens sont souvent incapables de dépister rapidement un trouble qui pourrait être traité, surtout lorsqu’il s’agit d’affections à apparition tardive ou d’affections légères, dont les symptômes ne se manifestent pas tous. Le rendement diagnostique du bilan métabolique habituel (lactate, ammoniaque, chromatographie des acides aminés plasmatiques et acides organiques urinaires) oscille entre moins de 1 % et 5 % [7]. Il n’est donc justifié qu’en présence de signes cliniques. Cependant, les études antérieures visaient à établir une étiologie, sans s’attarder au « rendement thérapeutique » (c’est-à-dire le dépistage d’une affection traitable). Dans une étude faisant appel à un bilan métabolique plus étendu, la capacité diagnostique était supérieure à 5 % [5]. On sait aussi que de nombreuses affections qui, au départ, ne se manifestent pas par une régression du développement sont des causes traitables de RGD et de HI [5]. Le BC Children’s Hospital [5] a lancé une initiative canadienne qui découle d’une analyse bibliographique [16][17] recensant 89 EIM traitables [17]. Cette initiative vise à dépister les maladies avant qu’elles deviennent graves ou que des complications irrémédiables se déclarent. Le protocole repose sur un algorithme à deux niveaux. Le niveau 1 est formé d’un groupe de tests qui permettent de dépister au moins trois EIM et qui sont facilement accessibles, peu invasifs et peu coûteux (à Vancouver, ces tests coûtent 528 $).
Les tests de niveau 1 peuvent dépister 60 % des EIM traitables qu’on sait responsables d’un HI. L’application TreatableID.org (www.treatable-id.org), offerte en anglais, contient un algorithme qui est régulièrement mis à jour et qui contient 81 HI traitables décrits par leurs anomalies biochimiques, les tests diagnostiques qui s’y associent, leurs caractéristiques cliniques et leurs modalités thérapeutiques [17]. Les développeurs de l’algorithme recommandent d’effectuer les tests de niveau 1 avant les tests génétiques et la neuro-imagerie. Ils soulignent la capacité de traiter les affections présentées dans l’algorithme et l’urgence relative de les dépister. L’AAP recommande d’effectuer un bilan métabolique soit conjointement avec l’ACM et le test d’ADN du gène de l’X fragile, soit peu après [2]. Les deux groupes proposent les mêmes tests au niveau 1, sauf que le protocole TIDE ajoute le dosage du cuivre et de la céruloplasmine. Quant à l’AAN, elle ajoute des tests métaboliques de base qui peuvent orienter la tenue d’autres tests : glycémie, gaz sanguins, lactate et créatine kinase. Au tableau 5 et à la figure 1 sont résumées les explorations de laboratoire de niveau 1 à effectuer chez tous les patients dont le RGD ou le HI ne s’accompagne pas d’une constellation de symptômes reconnaissable. En présence de certains signes évocateurs, il faut envisager de demander une évaluation à un spécialiste du métabolisme ou discuter du cas avec lui afin d’adapter les explorations de laboratoire en fonction du patient.
D’AUTRES EXPLORATIONS
La fonction thyroïdienne
L’hypothyroïdie est une cause courante et réversible de RGD et de HI. Son incidence est d’environ un cas sur 3500 naissances vivantes. De nombreux auteurs recommandent d’effectuer un bilan de dépistage de la fonction thyroïdienne [9], mais selon l’AAN, il n’est pas nécessaire de le reprendre après un dépistage néonatal [4]. Ce test fait partie des explorations de niveau 1 (tableau 5), qu’un dépistage néonatal ait été effectué ou non, afin de ne pas rater les cas acquis et les hypothyroïdies d’origine hypothalamique ou pituitaire.
Le fer, la vitamine B12
Un groupe australien [9] recommande d’inclure l’hémogramme et le dosage de la ferritine et de la vitamine B12 dans le bilan initial des enfants atteints de RGD ou de HI, particulièrement en présence d’une anamnèse de pica ou de restrictions alimentaires. L’anémie par carence en fer est une cause de perturbations du développement qui est facile à dépister et à soigner.
Le dosage du plomb
L’intoxication par le plomb peut nuire gravement au déve loppement mental et physique, particulièrement chez les enfants de moins de cinq ans, et provoquer des affections comme le trouble du spectre de l’autisme, la régression du développement (particulièrement en ce qui a trait au langage) et l’encéphalopathie [18]. L’AAN est la seule association à recommander le dosage du plomb chez les enfants présentant des facteurs de risque d’exposition.
Le dépistage d’infections congénitales
Une étude [9] propose d’évaluer les infections congénitales (TORCH : toxoplasmose, rubéole, CMV, herpès, autres) en présence d’anomalies neurologiques, de microcéphalie ou de perte de la vision ou de l’ouïe. Le clinicien doit envisager de consulter un infectiologue s’il craint une infection congénitale.
La neuro-imagerie
Les études de neuro-imagerie, y compris la tomodensitométrie et l’imagerie par résonance magnétique (IRM), révèlent des anomalies non spécifiques chez environ 30 % des enfants ayant un RGD ou un HI [6] (quelque part entre 2 % et 80 %, selon l’étude), mais elles contribuent à déterminer l’étiologie du RGD ou du HI dans seulement 0,2 % à 2,2 % des cas [2]. Le rendement diagnostique de la neuro-imagerie est plus positif en présence d’un examen neurologique anormal, de convulsions, de macrocéphalie ou de microcéphalie. L’IRM est préférable à la tomodensi tométrie, car elle est plus sensible pour dépister des anomalies structurelles importantes sur le plan clinique et des anomalies liées à la myélinisation et à la migration neuronale [2][4][9]. Puisque l’IRM exige souvent une sédation et que la découverte d’une anomalie donne rarement lieu à un diagnostic étiologique, l’AAP ne recommande pas systématiquement la neuro-imagerie chez les enfants ayant un RGD ou un HI. L’AAN la recommande chez tous les patients dont l’ACM, le test d’ADN du gène de l’X fragile et l’analyse du gène MECP2 (s’il y a lieu) ne sont pas concluants [4]. D’autres la recommandent seulement en présence de manifestations neurologiques particulières [9]. Selon l’avis d’experts, l’IRM du cerveau par spectroscopie est indiquée dans tous les cas d’épilepsie réfractaire ou de régression du développement.
L’électroencéphalographie
L’épilepsie ou les syndromes épileptiques non contrôlés, tels que le syndrome de Landau-Kleffner, peuvent être liés à des retards ou des régressions du développement. Les convulsions sont un symptôme courant des EIM. L’électroencéphalographie est justifiée en cas de présomption clinique de convulsions, de régression du langage ou de trouble neurodégénératif [9].
Un algorithme pour effectuer les tests
Une approche graduelle, fondée sur le document de principes de 2014 de l’AAP, les directives de 2011 de l’AAN et le protocole TIDE, modifiés pour tenir compte des publications scientifiques et des consensus d’experts, est présentée à la figure 1.
SOMMAIRE
Le RGD et le HI sont des troubles courants chez les enfants, et les pédiatres participent souvent au bilan étiologique nécessaire pour poser le diagnostic et déterminer les mesures à prendre. Même s’il est crucial de dépister rapidement le problème et d’entreprendre un programme de stimulation précoce, un diagnostic étiologique importe également, car il peut contribuer à soulager le stress de la famille, limiter le nombre de tests invasifs et inappropriés, orienter le pronostic et, dans certains cas, modifier la prise en charge et le traitement et prévenir les complications. De plus en plus, les tests visant le « rendement thérapeutique » sont préférés à ceux visant l’obtention d’un diagnostic pur dans ce domaine à l’évolution rapide. Les enfants chez qui on présume un RGD ou un HI profiteront à coup sûr des nouvelles approches exposées dans le présent document de principes.
RECOMMANDATIONS
Les recommandations suivantes reposent sur des directives cliniques fondées sur des données probantes et sur des avis d’experts :
- L’anamnèse et l’examen physique demeurent les meilleures premières mesures à prendre pour poser un diagnostic. Il faut les effectuer systématiquement auprès de chaque enfant ayant un retard global de développement (RGD) ou un handi cap intellectuel (HI) présumé. Lorsque l’évaluation clinique oriente les cliniciens vers un diagnostic particulier, il faut d’abord demander une exploration pour confirmer cette étiologie.
- Lorsque l’évaluation clinique n’oriente pas le clinicien vers un diagnostic précis, il faut envisager une approche graduelle de l’exploration. La portée de l’exploration dépendra de l’expérience du pédiatre, de l’accès aux surspécialistes et des ressources disponibles.
- Pour promouvoir l’évaluation des enfants ayant un RGD ou un HI selon une approche fondée sur des données probantes, il est essentiel que les centres spécialisés provinciaux, territoriaux ou régionaux qui effectuent les tests collaborent avec les médecins qui coordonnent les soins.
- Il est capital d’effectuer des tests de la vision et de l’ouïe en bonne et due forme chez tous les patients atteints d’un RGD ou d’un HI présumé.
- Lorsqu’aucun diagnostic étiologique n’est posé après l’anamnèse et l’examen physique, il est recommandé d’effectuer le test d’ADN du gène de l’X fragile, l’analyse chromosomique sur micropuce (hybridation génomique comparative, ou CGH) et les tests métaboliques de niveau 1, avec ou sans imagerie cérébrale. Si aucun dia gnostic n’est posé, il faut envisager de consulter un spécia liste de la génétique ou du métabolisme.
- L’analyse chromosomique sur micropuce et le test d’ADN du gène de l’X fragile sont des explorations de première ligne pour les enfants ayant un RGD ou un HI inexpliqué.
- Les données probantes appuient les tests de niveau 1 (tableau 5) pour dépister les erreurs innées du métabolisme traitables chez les enfants ayant un RGD ou un HI inexpliqué, même en l’absence de signes cliniques évocateurs et même lorsque le dépistage néonatal était normal.
- L’imagerie cérébrale est recommandée en première ligne chez les patients ayant une microcéphalie, une macrocéphalie, des convulsions ou des signes neurologiques anormaux. Autrement, l’imagerie peut être reportée après les explorations génétiques et métaboliques de première ligne. Dans chaque cas, il faut soupeser les risques et les avantages de la sédation. L’imagerie par résonance magnétique est la modalité de choix.
- Il faut vérifier le dosage du plomb et du fer chez les enfants à risque.
- Le séquençage de l’exome ou du génome entier pourrait être indiqué en milieu clinique lorsqu’il sera plus accessible.
Remerciements
Le comité de la pédiatrie communautaire et le groupe de travail de la petite enfance de la Société canadienne de pédiatrie ont révisé le présent document de principes.
COMITÉ DE LA SANTÉ MENTALE ET DES TROUBLES DU DÉVELOPPEMENT DE LA SOCIÉTÉ CANADIENNE DE PÉDIATRIE
Membres : Debra Andrews MD (présidente), Stacey A. Bélanger MD (présidente sortante), Alice Charach MD, Brenda Clark MD (membre sortante), Mark Feldman MD (représentant du conseil), Benjamin Klein MD, Daphne Korczak MD (membre sortante), Oliva Ortiz-Alvarez MD
Représentantes : Sophia Hrycko MD, Académie canadienne de psychiatrie de l’enfant et de l’adolescent; Angie Ip MD, section de la pédiatrie du développement de la SCP; Aven Poynter MD, section de la santé mentale de la SCP
Auteures principales : Stacey A. Bélanger MD Ph. D., Joannie Caron MD
Références
- American Psychiatric Association. Handicaps intellectuels. In Manuel diagnostique et statistique des troubles mentaux, 5e édition. Elsevier Masson SAS, Issy-les-Moulineaux: APA, 2015.
- Moeschler JB, Shevell M; Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 2014;134(3):e903–18.
- Jimenez-Gomez A, Standridge SM. A refined approach to evaluating global developmental delay for the international medical community. Pediatr Neurol 2014;51(2):198–206.
- American Academy of Neurology. Evaluation of the Child with Global Developmental Delay. 2011. www.aan.com/guidelines (consulté le 17 mars 2017).
- van Karnebeek CD, Stockler-Ipsiroglu S. Early identification of treatable inborn errors of metabolism in children with intellectual disability: The Treatable Intellectual Disability Endeavor protocol in British Columbia. Paediatr Child Health 2014;19(9):469–71.
- Shaffer LG; American College of Medical Genetics Professional Practice and Guidelines Committee. American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation. Genet Med 2005;7(9):650–4.
- Tirosh E, Jaffe M. Global developmental delay and mental retardation–A pediatric perspective. Dev Disabil Res Rev 2011;17(2):85–92.
- Wong VC, Chung B. Value of clinical assessment in the diagnostic evaluation of global developmental delay (GDD) using a likelihood ratio model. Brain Dev 2011;33(7):548–57.
- Silove N, Collins F, Ellaway C. Update on the investigation of children with delayed development. J Paediatr Child Health 2013;49(7):519–25.
- Manning M, Hudgins L; Professional Practice and Guidelines Committee, American College of Medical Genetics. Array-based Technology and Recommendations for Utilization in Medical Genetics Practice for Detection of Chromosomal Abnormalities. 2010. www.acmg.net/StaticContent/PPG/Array_based_technology_and_recommendations_for.13.pdf (consulté le 17 mars 2017).
- Sherr EH, Michelson DJ, Shevell MI, Moeschler JB, Gropman AL, Ashwal S. Neurodevelopmental disorders and genetic testing: current approaches and future advances. Ann Neurol 2013;74(2):164–70.
- Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol 2012;19(4):173–80.
- Hersh JH, Saul RA; Committee on Genetics. Health supervision for children with Fragile X syndrome. Pediatrics 2011;127(5):994–1006.
- Wright CF, McRae JF, Clayton S et coll. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med 2018.
- Boycott K, Hartley T, Adam S et coll.; Collège canadien des généticiens médicaux. The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position statement of the Canadian College of Medical Geneticists. J Med Genet 2015;52(7):431–7.
- van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol Genet Metab 2012;105(3):368–81.
- van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: Diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab 2014;111(4):428–38.
- Lowry JA. Childhood Lead Poisoning: Clinical Manifestations and Diagnosis. 2016. www.uptodate.com/contents/childhood-lead-poisoning-clinical-manifestations-and-diagnosis?source=search_result&search=Childhood+lead+poisoning&selectedTitle=1%7E150 (consulté le 17 mars 2017).
Avertissement : Les recommandations du présent document de principes ne constituent pas une démarche ou un mode de traitement exclusif. Des variations tenant compte de la situation du patient peuvent se révéler pertinentes. Les adresses Internet sont à jour au moment de la publication.
Mise à jour : le 7 février 2024